Dabble¶
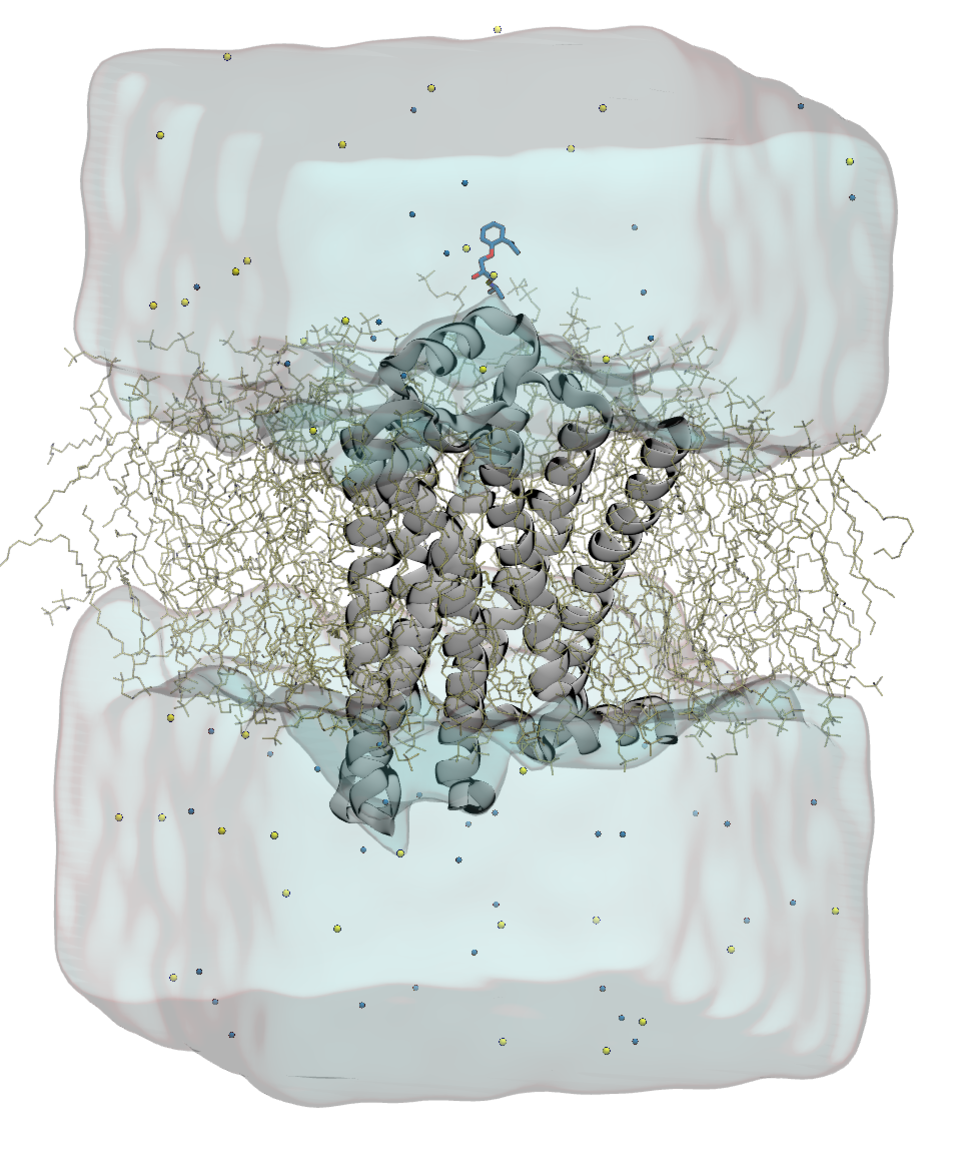
Dabble is a tool for building membrane protein systems. The ultimate goal of the project is to create an easy to use, one stop tool for system construction and parameterization.
Dabble includes both the main program dabble.py as well as a powerful API for building and parameterizing molecular dynamics systems.
To get started via Anaconda Python, use:
conda install -c rbetz dabble
Features¶
Prepare membrane protein systems by inserting them into a membrane
Prepare solvated proteins by adding water
Add ions to neutralize and/or to desired concentration
Parameterize with AMBER, CHARMM, or GROMACS format parameter sets
Outputs files for simulation with most major simulation codes
Automatic detection of post-translational modifications
Modified amino acids made easy
Ligands made easy! No more messing with atom names.
Supported Parameter Sets¶
[X] AMBER
[X] CHARMM
[X] OPLS-AA (no lipids)
[X] Gromacs (no covalent modifications)
Supported Water Models¶
[X] TIP3
[X] TIP4P-EW
[X] SPC/E
Supported Simulation Programs¶
[X] AMBER
[X] Anton (via conversion)
[X] CHARMM
[X] Desmond
[X] GROMACS
[X] LAMMPS
[X] NAMD
[X] OpenMM
[X] Any code accepting .psf, .prmtop, or .gro
Citing Dabble¶
There is no paper on Dabble yet. If you use Dabble, please cite it with the following DOI:
Coming soon¶
Dabble is under active development. The following features are coming soon:
Support for covalent modifications in GROMACS
Lipids with OPLS-AA parameters
TIP4P-EW waters with CHARMM parameters, in .prmtop format
Contributing¶
Dabble is a written by Robin Betz. Bug finding is always appreciated, as well as corner cases where your protein won't dabble.
Dabble is licensed under the GPLv2 license.
A lot of Dabble is powered by the VMD Python API.
Source code: https://github.com/drorlab/dabble/
Issue tracker: https://github.com/drorlab/dabble/issues